Ass. Prof. Dr. Michael Haumann
Biophysics of Metalloenzymes

Welcome!
Room 1.2.23, Tel.: +49 (0)30 838 56101
14195 Berlin, Germany
See my pages "Teaching" for information on courses in the winter semester 2025/26!
Diesem Link bitte zu unserer deutschen Titelseite folgen 
Nature has recruited metal ions in a fascinating assortment of biological systems. About half of all proteins contain metal ions. Remarkably, metalloenzymes show high rates for small molecule conversion reactions (H2O, H2, O2, CO, CO2, CH4), which are outstandingly important, for example, in renewable energy, medical, and industrial catalysis applications.
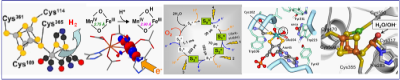
We focus on basic research on biological metal centers and biomimetic model complexes in small molecule catalysis. Molecular structures, electronic configurations, and kinetics of intermediate formation are characterized to unravel, for example, the cofactor assembly and substrate turnover mechanisms. Generalized insights are derived and employed to understand the restraints that govern metal-centered (bio)catalysis.

A broad range of enzymes and models is studied by us, in collaboration with worldwide researchers. Hydrogenases form H2 at Fe and Ni centers and are promising for future energy applications. Various oxidases contain prototypic binuclear Fe and Mn sites, involved in O2 activation and CH4 conversion. Photosynthetic water oxidation and atmospheric O2 formation at the Mn-complex of photosystem II is a key to solar fuels. Mo-enzymes catalyze numerous reactions in the cell. CO/CO2 conversion by Ni-Fe enzymes is a new topic in this group.

Our central tool is X-ray spectroscopy at synchrotron radiation sources (ANKA, BESSY, DESY, ESRF, MAXLAB, SLS, SOLEIL). We use X-ray absorption spectroscopy (XANES, EXAFS), X-ray emission spectroscopy (XES), resonant inelastic X-ray scattering (RIXS), and nuclear resonance vibrational spectroscopy (NRVS). Thereby, metal site structures and electronic configurations are determined at the atomic level, with µm spatial resolution, and µs time resolution. Density functional theory calculations (DFT) are used to obtain optimized structural models, spectral simulations, and insights into electronic structures.