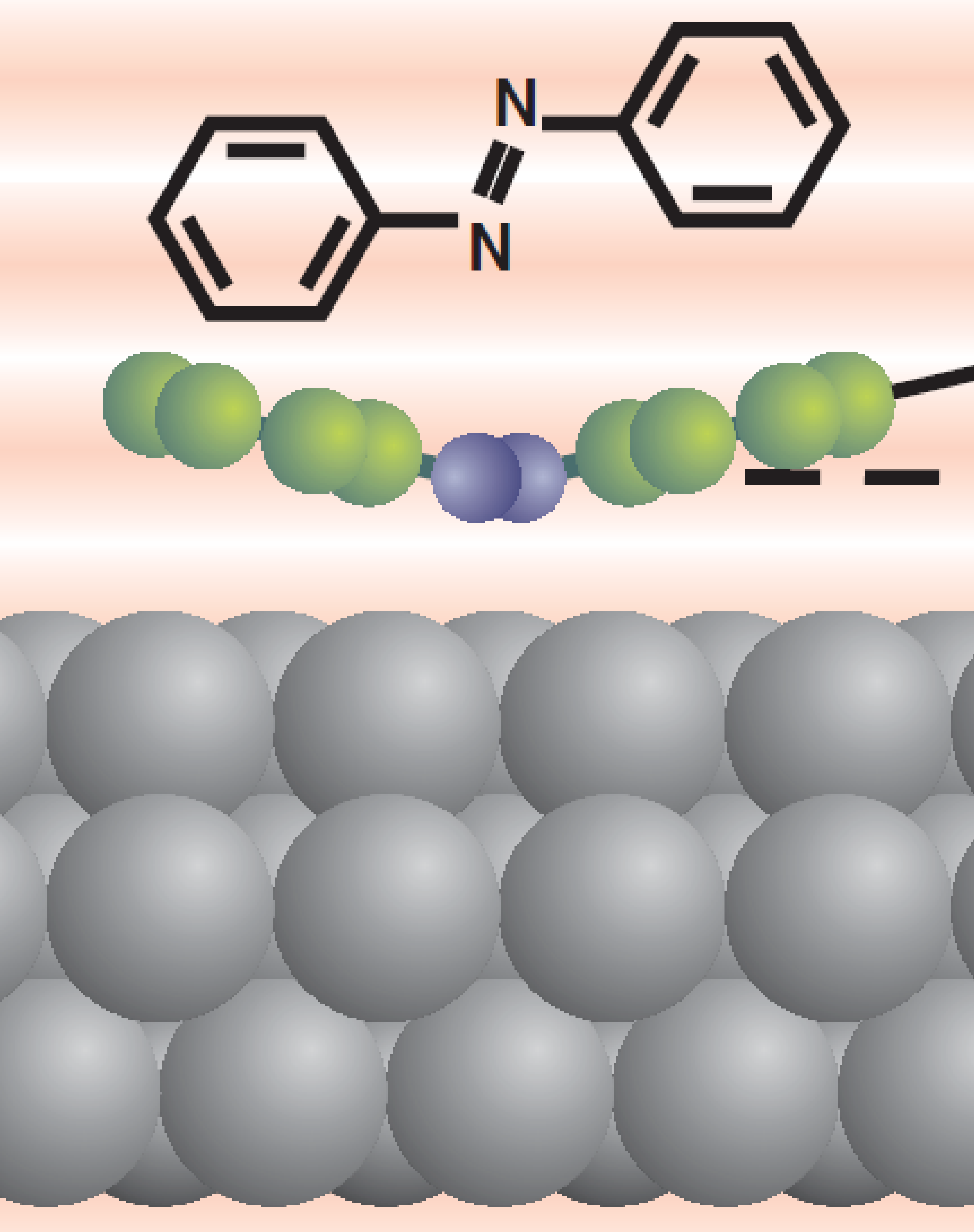
Abstract.We employ normal-incidence x-ray standing wave and temperature programed desorption spectroscopy to derive the adsorption geometry and energetics of the prototypical molecular switch azobenzene at Ag(111). This allows us to assess the accuracy of semiempirical correction schemes as a computationally efficient means to overcome the deficiency of semilocal density-functional theory with respect to long-range van der Waals (vdW) interactions. The obtained agreement underscores the significant improvement provided by the account of vdW interactions, with remaining differences mainly attributed to the neglect of electronic screening at the metallic surface.
Abstract.Graphene as a zero-bandgap semiconductor is an ideal model structure to study the carrier relaxation channels, which are inefficient in conventional semiconductors. In particular, it is of fundamental interest to address the question whether Auger-type processes significantly influence the carrier dynamics in graphene. These scattering channels bridge the valence and conduction band allowing carrier multiplication, a process that generates multiple charge carriers from the absorption of a single photon. This has been suggested in literature for improving the efficiency of solar cells. Here we show, based on microscopic calculations within the density matrix formalism, that Auger processes do play an unusually strong role for the relaxation dynamics of photoexcited charge carriers in graphene. We predict that a considerable carrier multiplication takes place, confirming the potential of graphene as a new material for high-efficiency photodevices.
M. S. Alam, M. Stocker, K. Gieb, P. Müller, M. Haryono, K. Student, and A. Grohmann
Abstract.Strung out: FeII complexes containing pairs of planar terdentate N ligands form regular one-dimensional aggregates on highly oriented pyrolytic graphite, on a length scale of hundreds of nanometers. STM spectroscopy was used to probe the molecular conductance and thus the spin state. The distribution of spin states is random in the single-molecule chain, but local cooperativity sets in when the chain is made up of oligonuclear “beads”.
C. Gahl, R. Schmidt, D. Brete, E. R. McNellis, W. Freyer, R. Carley, K. Reuter, and M. Weinelt
Abstract.Optical properties and the geometric structure of self-assembled monolayers of azobenzene-functionalized alkanethiols have been investigated by UV/visible and near edge X-ray absorption fine structure spectroscopy in combination with density-functional theory. By attaching a trifluoro-methyl end group to the chromophore both the molecular tilt and twist angle of the azobenzene moiety are accessible. Based on this detailed structural analysis the energetic shifts observed in optical reflection spectroscopy can be qualitatively described within an extended dipole model. This substantiates sizable excitonic coupling among the azobenzene chromophores as an important mechanism that hinders trans to cis isomerization in densely packed self-assembled monolayers.